What we learn by CRISPR'ing a random set of regulatory elements
What can we learn about the properties of gene regulatory elements by CRISPR’ing a random set of accessible sites in human cells?

We and others have previously used CRISPRi to perturb thousands of candidate regulatory elements and measure their effects on expression. Those studies have taught us a lot about how regulatory elements work, and they let us build predictive models like ABC and ENCODE-rE2G. But the datasets they rely on carry some key selection biases that could skew our view of what regulatory elements actually look like:
- They often selected “interesting” elements (for example, sites with high H3K27ac) or interesting genes (for example, transcription factors).
- They largely focused on a single cell type — K562 cells.
- Statistical power was limited by cost.
To get around this, we did three things. We used CRISPRi to perturb about 1,000 randomly selected elements across 25 loci in each of two cell types. We developed DC-TAP-seq to improve guide capture and get high capture for the genes we care about. And we built a statistical power framework to make sure we were actually powered to detect 15–25% effects on gene expression.
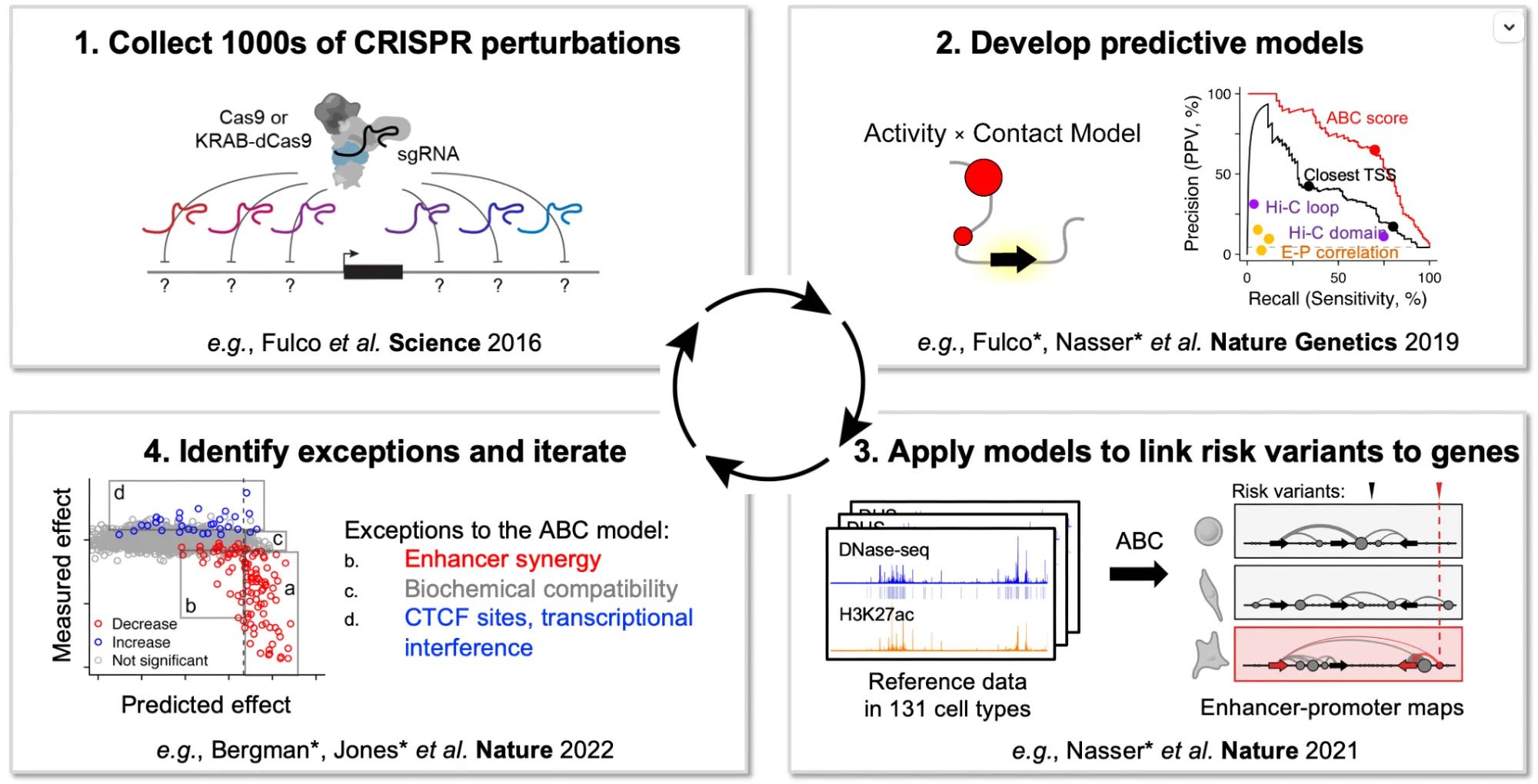
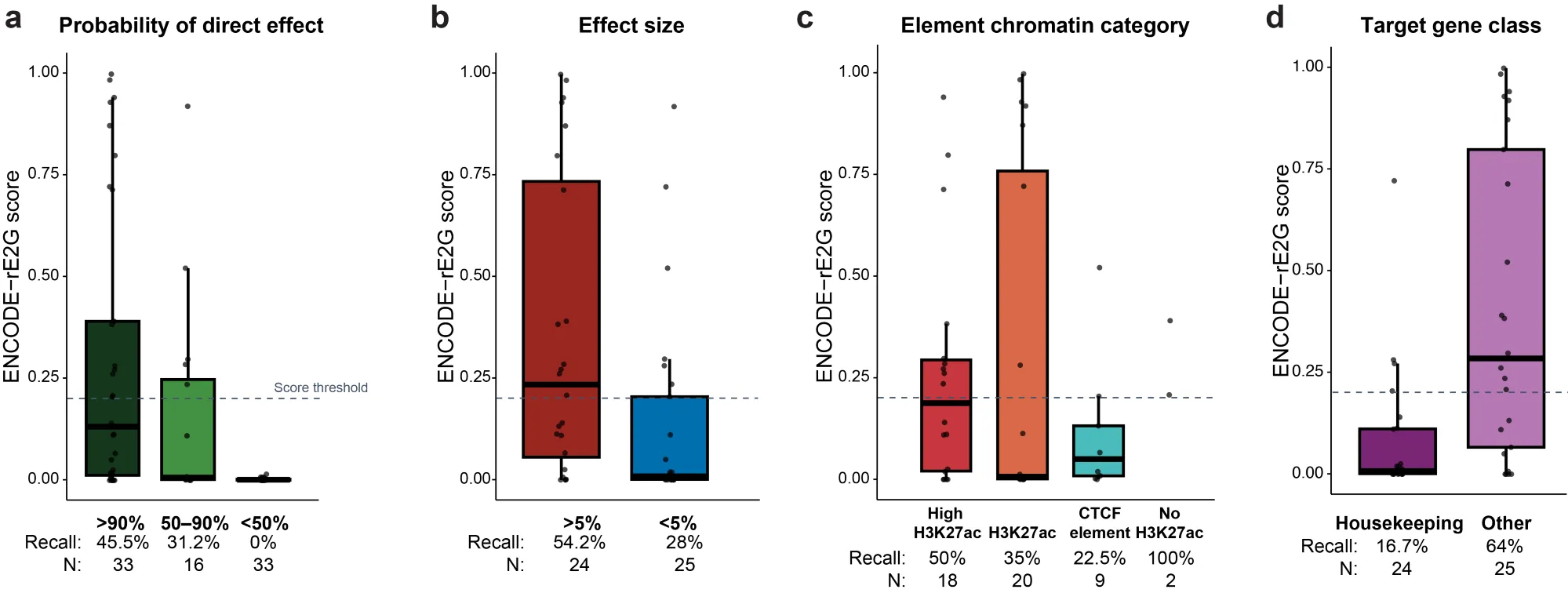
We found 145 significant element–gene pairs, out of 4,711 tested with good power for 15% effect sizes — and the properties of these interactions differed from previous studies in a few important ways.
Most effect sizes fell in the range of 5–10% — much smaller than the effects reported in earlier studies. This wasn’t a technical artifact; it reflects differences in statistical power and in how elements and genes were selected.
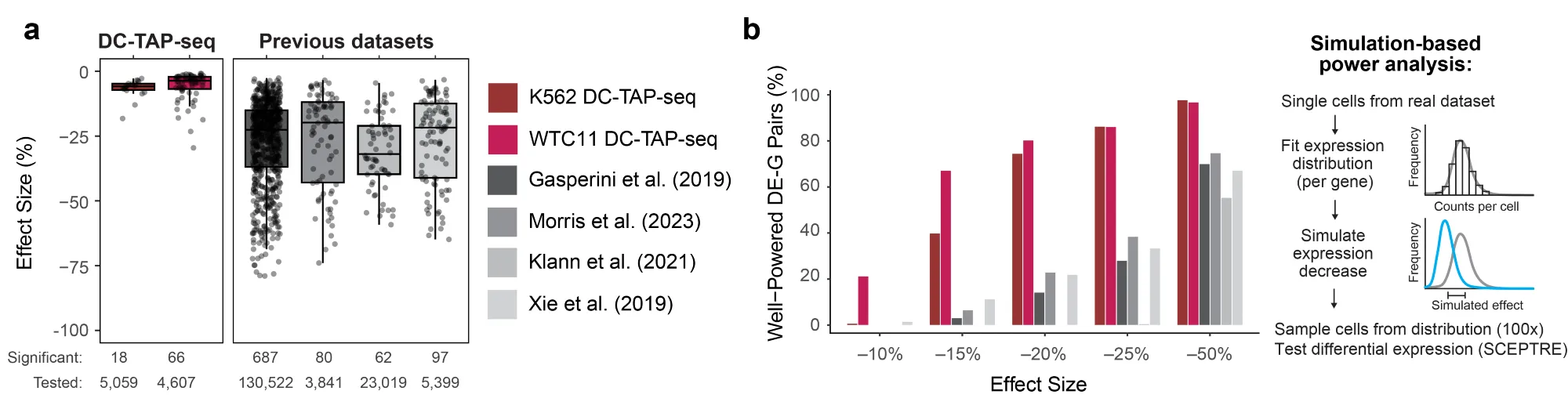
Nearly half of the significant effects were likely to be indirect — including nearly all of the apparent cases of “up-regulation.” So CRISPRi is not, for example, turning up lots of silencing elements.
Seventeen percent of the regulatory elements corresponded to sites that bind CTCF only, with no or very low H3K27ac. These sites — likely CTCF binding sites that regulate 3D contacts — are common, and they’ve been missed in some previous studies precisely because those studies selected elements with high H3K27ac.
Housekeeping genes appeared to have a similar frequency of distal enhancers as non-housekeeping genes, but the effects of those enhancers were roughly 2-fold weaker. That’s consistent with earlier work suggesting the promoters of housekeeping genes are less responsive to distal enhancers.
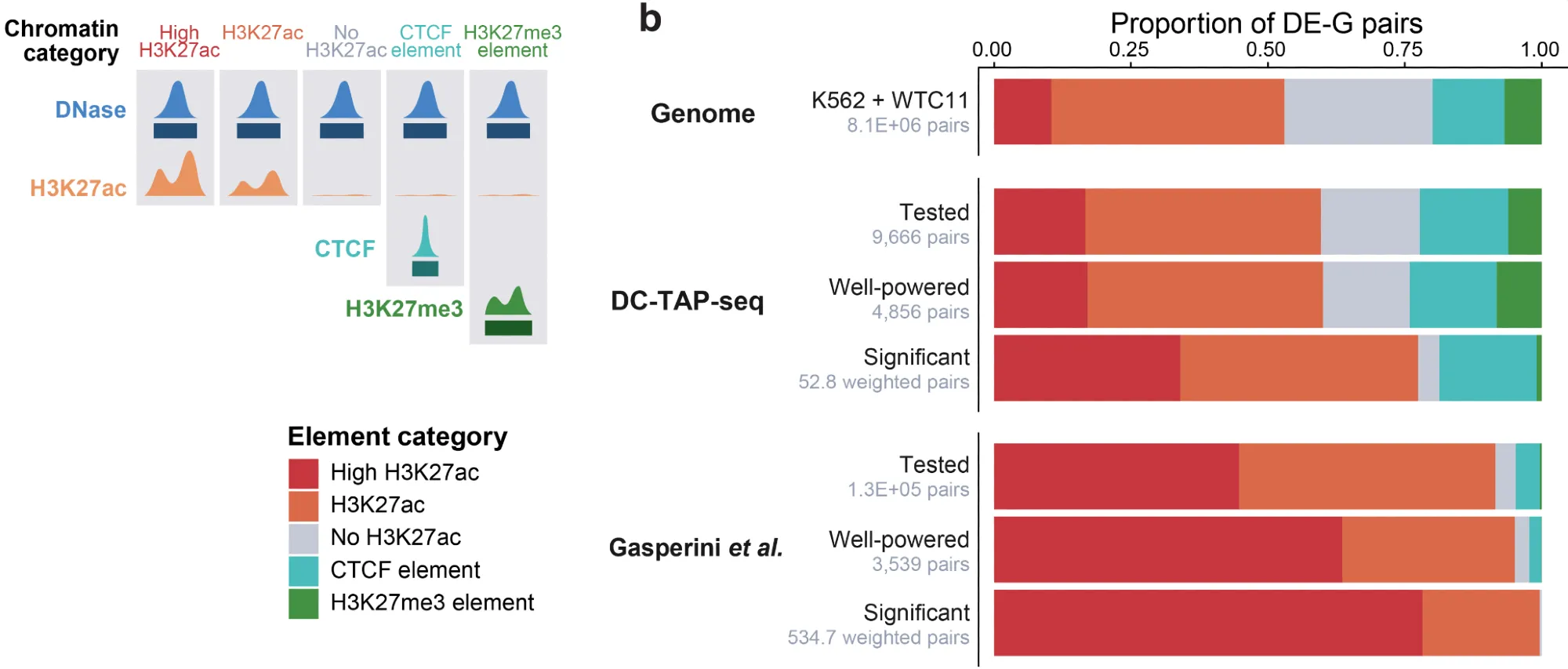
These unbiased datasets are exactly what we need to evaluate predictive models (stay tuned for results on scE2G and ENCODE-rE2G). One lesson already: any such evaluation has to account for the magnitude of effect sizes, the frequency of indirect effects, chromatin state, and gene class. Reassuringly, the observations held across both cell types (K562 and hiPSCs), suggesting they generalize beyond the favorite workhorse cancer cell line.
We hope these tools are useful to you. This study is our most complete toolkit to date for designing, running, and analyzing regulatory-element CRISPR perturbation studies — code, protocols, and data are all available now. We’d love to help you set up these experiments in new systems, and to expand these data 10- to 100-fold over the next few years to better understand regulatory elements, improve predictive models, and interpret genetic variants. (As one example, the Quertermous lab has already used DC-TAP-seq to target elements containing coronary-artery-disease variants in smooth muscle cells — the high statistical power will be important for finding effects in GWAS loci.)
This was a huge team effort. Judhajeet, Evvie, and Dulguun led the development of DC-TAP-seq and the design and execution of the random screens; James and Evvie led the analysis; and Maya and Andreas compared the effects to models and teased out the indirect effects. Congratulations, all.
Read the preprint · Adapted from the original thread on Bluesky.